Responsible person: Assoc. Prof. Jaroslav Roh
Tuberculosis (TB) is a widespread infectious disease and presents the second greatest killer worldwide due to a single infectious agent. About one third of world population has latent form of TB, which can be activated mainly during immunosuppression (e.g. HIV infection). In 2019, 10 mil new cases of TB were found and 1.4 mil people died from TB. Although tuberculosis is curable and preventable disease, the poor quality medicine, incorrect treatment as well as low availability of the medication cause serious problems in the therapy and lead to the formation of resistant strains. Multidrug-resistant TB (MDR-TB) is caused by strains resistant to isoniazid and rifampicin, the most powerful antituberculosis drugs, with or without resistance to other first-line drugs. Extensively drug-resistant tuberculosis (XDR-TB) arose from MDR strains and exhibits an added resistance to fluoroquinolones and second-line injectable aminoglycosides (kanamycin, amikacin and capreomycin). In 2019, WHO estimates that there were 465 000 new MDR-TB or rifampicin-resistant TB cases. Worldwide, only 57% of MDR-TB patients are successfully cured.
Source: World Health Organization – Tuberculosis; http://www.who.int/topics/tuberculosis/en/
Therefore, the search for new antituberculosis drugs, active also against these resistant strains, is highly important. Our group deals with the synthesis of new antituberculotics, with structures different from known anti-TB drugs and with expected novel mechanisms of action.
General objective
Our research group deals with the synthesis of nitrogen-containing heterocyclic derivatives with potential antimycobacterial activity. Antimycobacterial activitites of our compounds are evaluated in vitro against collection strains of Mycobacteria sp. and also agains multi-drug resistant strains of Mycobacterium tuberculosis isolated from patients. In order to determine the selectivity, effect of our compounds on human cell lines and their activities against bacterial and fungal strains are also studied.
The compounds with the most promising results are further studied in vivo. Firstly, we determine some basic pharmacokinetic parameters. Then, antituberculosis action of the selected compounds are studied in vivo in tuberculosis mouse model at the Biological Defence Department at Těchonín.
Main results since 2014
In the last decade, series of compounds based on 2-benzylsulfanylbenzimidazoles, 2-benzylsulfanylbenzoxazoles, 2-benzylsulfanylbenzothiazoles as well as 3-benzylsulfanyltriazoles and 2- or 4-benzylsulfanylpyridines were synthesized and studied. Especially benzazole derivatives bearing dinitrobenzylsulfanyl fragment exhibited antimycobacterial activities comparable to isoniazid, first-line antituberculosis drug.
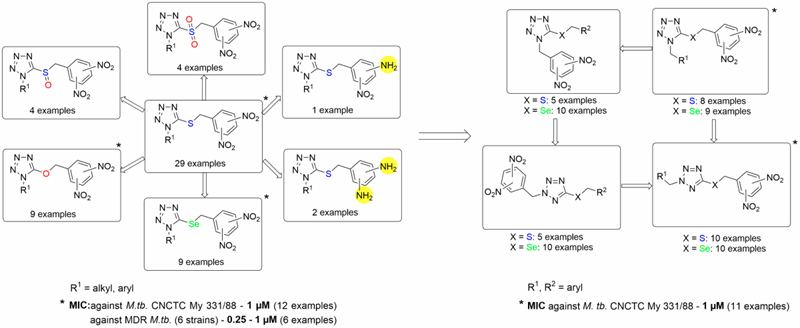
Later on, we reported a huge structure-activity relationship study of dinitrobenzylsulfanyl tetrazoles and their analogs. As a result of this study, 1-substituted-5-(3,5-dinitrobenzylsulfanyl)-1H-tetrazoles and their oxa and selenium analogs have been identified as the new lead compounds. They displayed high and selective antimycobacterial activity, low cellular toxicity and genotoxicity. Furthermore, they are active also against MDR M. tuberculosis strains, with no cross-resistance with currently used antiTB drugs (Figure 1).
Moreover, 2,5-regioisomeric analogs of lead compounds - 2-substituted-5-(3,5-dinitrobenzylsulfanyl)-2H-tetrazoles - have more beneficial properties compared to 1,5-regioisomers (Figure 1).
G. Karabanovich et al. EJMC 2014 G. Karabanovich et al. MedChemComm 2015
Figure 1. Structure-activity relationships in the group of 3,5-dinitrobenzylsulfanyl tetrazoles and their analogs
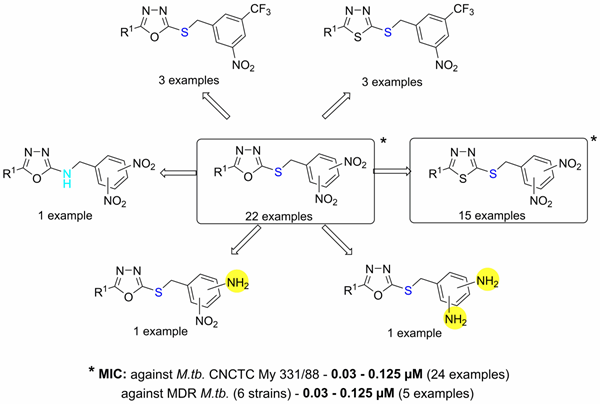
In 2016, we reported a structure-activity relationship study of dinitrobenzylsulfanyl oxadiazoles and thiadiazoles. Compounds described in this work possess outstanding and highly selective in vitro antimycobacterial activity against both replicating and nonreplicating mycobacteria (Figure 2). Several experiments suggested that these compounds possess a novel mechanism of action affecting the synthesis of mycobacterial nucleic acids.
G. Karabanovich et al. JMC 2016
Figure 2. Structure-activity relationships in the group of antitubercular 3,5-dinitrobenzylsulfanyl oxadiazoles and their analogs
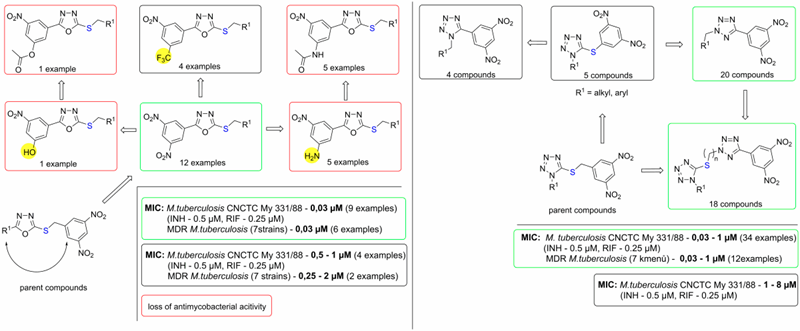
In 2017, we prepared a series of so called reverse analogs with 3,5-dinitrofenyl group attached directly to 1,3,4-oxadiazole of tetrazole. Surprisingly, these compounds showed excellent antimycobacterial activities comparable to those of 3,5-dinitrobenzylsulfanyl oxadiazoles parent compounds. Again, these compounds had highly selective action, as they were not active against other bacteria or fungi and had low effect on mammalian cell proliferation (Figure 3). Furthermore, series of 1- and 2-substituted 5-(3,5-dinitrophenyl)tetrazoles with high antimycobacterial activities were prepared and their structure-activity relationships were studied (Figure 3).
G. Karabanovich et al., EJMC 2017 J.Němeček et al., EJMC 2017
Figure 3. Structure-activity relationships in the group of antitubercular 3,5-dinitrophenyl oxadiazoles and tetrazoles
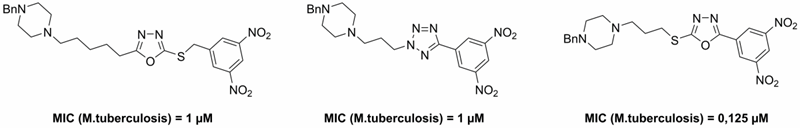
Then we focused on the water-soluble analogs of above described lead compounds. We modified the side chain of these highly active substances and introduced a tertiary amine-containing group. The final compounds was converted to water-soluble hydrochloride salts. The final hydrochlorides showed good solubility, in vitro antimycobacterial activity and selectivity towards mycobacteria (Figure 4).
J. Roh et al., BMC 2017
Figure 4. Benzylpiperazine-containing derivatives with good aqueous solubility and in vitro antimycobacterial activity
In 2019, several series of 3,5-dinitrophenyl containing triazoles were prepared and their antimycobacterial activities were described. Among the prepared compounds, the highest in vitro antimycobacterial activities against M. tuberculosis H37Rv and against seven clinically isolated multidrug-resistant strains of M. tuberculosis were found with S-substituted 4-alkyl-5-(3,5-dinitrophenyl)-4H-1,2,4-triazole-3-thiols and their 3-nitro-5-(trifluoromethyl)phenyl analogues. The minimum inhibitory concentrations of these compounds reached 0.03 μM, which is superior to all the current first-line anti-tuberculosis drugs. The docking study indicated that these compounds acted as the inhibitors of decaprenylphosphoryl-β-D-ribofuranose 2′-oxidase enzyme, which was experimentally confirmed by two independent radiolabeling experiments. (Figure 5)
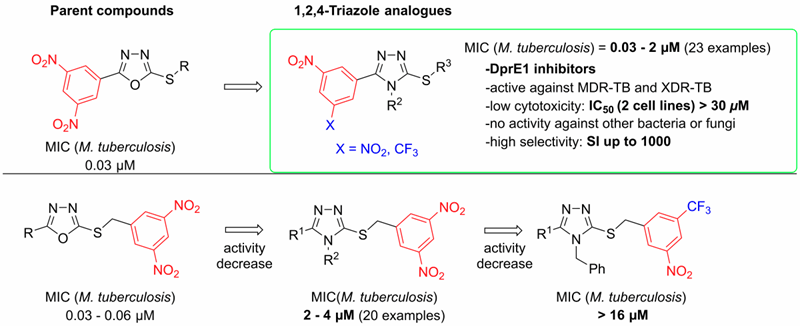
Figure 5. SAR of 3,5-dinitrophenyl containing 1,2,4-triazoles (G. Karabanovich et al. JMC 2019)