| This research project started with the foundation of Charles University Research Centre (UNCE 204019/304019/2012): Centre for the Study of Toxic and Protective Effects of Drugs on Cardiovascular System. This center officialy finished in decemebr 2017, but the cooperation between the groups involved in this centre continues. |
 |
Responsible person: Assoc. Prof. Jaroslav Roh
Iron chelating agents have been long used to the treatment of the iron overload diseases. However, in recent years they have also been thouroughly studied for their other effects such as antiproliferative, cardio- and neuro-protective activities. Our research group has been engaged in a development of aroylhydrazone chelators as cardioprotective and antiproliferative agents. In the field of cardioprotection we synthesize the derivatives of dexrazoxane and its metabolite ADR-925.
Synthesis of dexrazoxane analogues as potential cardioprotective agents
Anthracycline antineoplastic antibiotics such as doxorubicin or daunorubicin are widely used anticancer drugs. However, the administration of anthracyclines is connected with high risk of cardiotoxicity. Chronic anthracycline cardiotoxicity is characterized by dilated cardiomyopathy, with subsequent development of left ventricular contractile dysfunction and congestive heart failure. It is supposed that the complexation of anthracyclines with intracellular iron leads to the formation of reactive oxygen species, which causes serious tissue damage especially in myocardium. The only clinically used drug preventing anthracycline cardiotoxicity is dexrazoxane (DXZ). It was argued, that its mechanism of action involves iron-chelating properties of ADR-925, main metabolite of DXZ. However, our recent studies showed that the mechanism of action is more complex and involved the interaction of DXZ with beta isoforms of topoisomerase II (TOP2B) in heart.
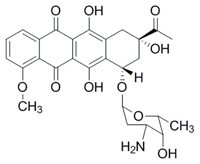
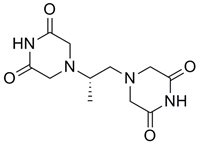
Daunorubicin Dexrazoxane
Recent results
From the very beginning of our research efforts, we focused on the role of a metabolite ADR-925 in the cardioprotective effect of DXZ. We developed the large-scale method for the preparation of the racemic form of ADR-925 and prepared more than 150 g of this substance (Figure 1). This was then used in advanced in vivo experiments on the model of chronic ANT cardiotoxicity in rabbits. The result of this long-lasting study is the reversal of the traditional hypothesis about the efficacy of dexrazoxane through its chelating metabolite ADR-925. The first part of this study, which focuses on the pharmacokinetics of dexrazoxane and its metabolite, was published in 2018.
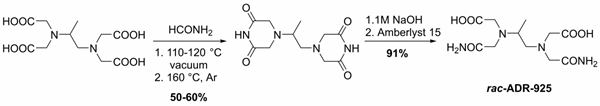
E. JIrkovský et al. JPET 2018
Figure 1. Large-scale synthesis of racemic form of ADR-925.
The second part of the study was focused on the pharmacodynamics of dexrazoxane and its metabolite ADR-925. We found, that unlike dexrazoxane, ADR-925 had no significant impact on daunorubicin-induced mortality, blood congestion, biochemical and functional markers of cardiac dysfunction in vivo. At the same time, dexrazoxane, but not ADR-925, inhibited and depleted TOP2B and prevented daunorubicin-induced genotoxic damage. As the result, our study strongly supports a new mechanistic paradigm that attributes clinically effective cardioprotection against anthracycline cardiotoxicity to interactions with TOP2B, but not metal chelation and protection against direct oxidative damage (E. Jirkovsky et al. Circulation Heart Failure 2021).
In 2020, we prepared several close analogues of dexrazoxane. However, all studied modifications, including simple methylation, were found to abolish the cardioprotective effects (Figure 2). As this challenged the prevailing mechanistic concept and previously reported data, the two closest derivatives (GK-627 and GK-580) were thoroughly scrutinized in vivo using a rabbit model of chronic anthracycline cardiotoxicity. In contrast to dexrazoxane, both compounds failed to protect the heart as demonstrated by mortality, cardiac dysfunction, and myocardial damage parameters, although the pharmacokinetics and metal-chelating properties of their metabolites were comparable to those of dexrazoxane. The loss of cardiac protection was shown to correlate with their abated potential to inhibit and deplete Top2B both in vitro and in vivo.
.jpg.aspx?lang=en-GB&width=500&height=234)
P. Kollárová-Brázdová et al. JPET 2020
Figure 2. Investigation of structure–activity relationships of dexrazoxane; structures of the studied analogs
In 2021, we evaluated a series of dexrazoxane analogues and found that their cardioprotective activity strongly correlated with their interaction with TOP2B in cardiomyocytes, but was independent of their iron chelation ability. We demonstrated very tight structure-activity relationships on stereoisomeric forms of 4,4'-(butane-2,3-diyl)bis(piperazine-2,6-dione). In contrast to its rac-form 12, meso-derivative 11 (ICRF-193) showed favorable binding mode to topoisomerase II in silico, inhibited and depleted TOP2B in cardiomyocytes more efficiently than dexrazoxane and showed the highest cardioprotective efficiency (Figure 3). Importantly, the observed ICRF-193 cardioprotection did not interfere with the antiproliferative activity of anthracycline. Hence, we identifies ICRF-193 as the new lead compound in the development of efficient cardioprotective agents.
.tif")
A. Jirkovska et al. J. Med. Chem. 2021
Figure 3. Identification of ICRF-193 as the new lead compound in the development of efficient cardioprotective agents
However, the poor aqueous solubility of ICRF-193 has precluded its further in vivo development as a cardioprotective agent. To overcome this issue, we developed several water-soluble prodrugs of ICRF-193 and demonstrated their sufficient solubility, low cytotoxicity and favorable ICRF-193 release in vivo. The treatment with the lead prodrug GK-667 was well tolerated and provided full protection against DAU-induced mortality and left ventricular dysfunction. Markers of cardiac damage/dysfunction revealed minor cardiac damage in the group co-treated with GK-667 in the lower dose, whereas almost full protection was achieved with the higher dose. This was associated with similar prevention of DAU-induced dysregulation of redox and calcium homeostasis proteins. GK-667 dose-dependently prevented p53-mediated DNA damage response in the LV myocardium not only in the chronic experiment but also after single DAU administration. These effects appear essential for cardioprotection, presumably due to the topoisomerase IIβ inhibition provided by its active metabolite ICRF-193. Hence, GK-667 merits advanced study as a promising drug candidate for cardioprotection against chronic ANT cardiotoxicity (Figure 4).

H. Bavlovič Piskáčková et al., Sci. Rep. 2021; P. Kollárová Brázdová et al., Clin. Sci. 2021; WO2021/144746 A1
Figure 4. Development of GK-667 as a promising drug candidate for cardioprotection against chronic ANT cardiotoxicity
Synthesis of hydrazone and thiosemicarbazone-type iron chelators
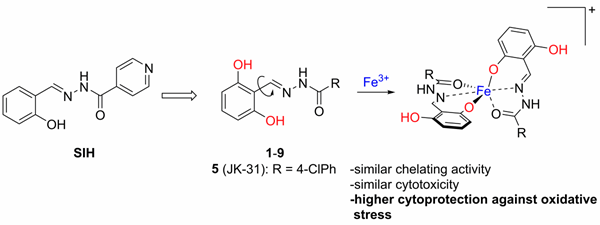
The hypothesis about the effectiveness of the chelating metabolite ADR-925 has led to our greater interest in iron chelators as potential cardioprotective agents but also more generally as protectants against oxidative stress and finally as effective antiproliferative agents. Hence we deals with the synthesis of hydrazone-type iron chelators derived from salicylaldehyde isonicotinoyl hydrazone (SIH) as cytoprotective agents.
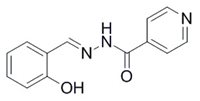
SIH
We are also involved in the research of thiosemicarbazone-type iron chelators as a potent antitumor agents. We prepared several semicarbazone and formamidrazone metabolites of these agents as a standards for pharmacokinetic studies.
In 2018, we prepared a series of 2,6-dihydroxybenzaldehyde analogues of SIH with increased hydrolytic stability and cytoprotective activity against oxidative stress compared to those of SIH (Figure 5). Compound JK-31 (2,6-dihydroxybenzaldehyde 4-chlorobenzohydrazone) showed the best cytoprotective efficiency among the studied compounds including SIH. This compound significantly protected H9c2 cardiomyoblast cells against oxidative stress induced by various pro-oxidants, such as hydrogen peroxide, tert-butyl hydroperoxide, paraquat, epinephrine, N-acetyl-p-benzoquinone imine (a toxic metabolite of paracetamol), and 6-hydroxydopamine.
H. Jansová et al. Chem. Res. Toxicol. 2018
Figure 5. 2,6-Dihydroxydihydroxybenzaldehyde analogues of SIH