Group of Cardiotoxicity
If you want to learn more abour our research, please see bottom of this page.
Group Leader
Postdoctoral Researchers
 |
Lenka Applová, Ph.D.
anthracycline cardiotoxicity
room 2324; applovl@faf.cuni.cz; +420 495 067 449
|
| |
|
 |
Hana Jansová, Ph.D.
oxidative stress-related cardiac injury, cardioprotective (pro)chelators of iron
room 2423; jansovah@faf.cuni.cz; +420 495 067 585
(maternity leave)
|
| |
|
 |
Anna Jirkovská, Ph.D. (maiden name Vávrová)
anthracycline cardiotoxicity, roles of DNA damage and topoisomerase II isoforms
room 2424; vavra3aa@faf.cuni.cz; +420 495 067 588
(maternity leave)
|
| |
|
Postgraduate (Ph.D.) Students
 |
Veronika Keresteš, M.Sc. (maiden name Skalická)
anthracycline cardiotoxicity
room 2353; skalicv1@faf.cuni.cz; +420 495 067 342
(maternity leave)
|
| |
|
 |
Kristína Moravová, M.Sc.
inhibitors of topoisomerase
room 2362; moravovkr@faf.cuni.cz; +420 495 067 297
|
| |
|
Most of our research projects aim at drug-induced cardiovascular toxicity, particularly the cardiotoxicity of anthracycline anticancer drugs, but we also examine cardiotoxic properties of other anticancer drugs (and/or their combinations with anthracyclines). Our next important topics are cardiac oxidative stress, catecholamine cardiotoxicity and the role of redox-active iron and other transition metals in oxidative stress and toxicity. We are engaged in several collaborative projects aimed at development of potential cardioprotective agents but also study novel potential anticancer agents with iron-chelating and photodynamic properties.


Isolation of the rat neonatal ventricular cardiomyocytes.
In most research topics, our in vitro cellular studies are coupled with experiments in collaborating laboratories using appropriate in vivo models and the pharmacodynamic / toxicodynamic data are often coupled with pharmaco-/toxico-kinetic studies. Predominantly, this collaborative research took place within the network of several groups joined in the Charles University Research Center for the Study of Toxic and Protective Effects of Drugs on Cardiovascular System. This center integrated approaches of biochemistry, medicinal chemistry, pharmacology, toxicology and bioanalytical chemistry, in order to bring together complementary skills, knowledge, and resources to jointly address the research problems.
<<< back on top
Anthracyclines (such as doxorubicin /syn. adriamycin/, daunorubicin or epirubicin) rank among the most effective anticancer drugs ever developed and have a broad spectrum of indications covering both hematological malignancies and solid tumors. However, their clinical utility is hampered by the risk of cardiotoxicity. The most troublesome is the chronic (or delayed) cardiotoxicity, which develops months or even years after the treatment and is typically characterized by progressive myofibrillar loss and degeneration of the left ventricular cardiomyocytes, ultimately resulting into dilated cardiomyopathy and congestive heart failure.
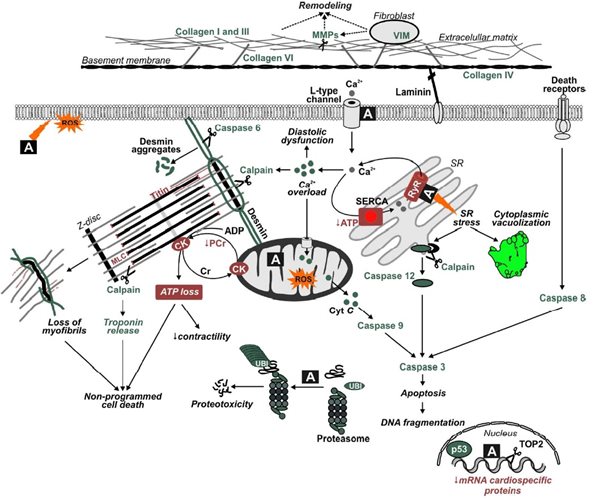
Despite a remarkably extensive literature on many aspects of anthracycline cardiotoxicity (see our recent comprehensive review), there is still insufficient understanding of its exact pathogenesis. Hence, in a close collaboration with our colleagues employing unique in vivo chronic animal model and modern molecular techniques,  we study the molecular mechanisms of anthracycline cardiotoxicity and particularly aim at advancement of understanding of the roles of redox-active iron, topoisomerase II and mitochodrial injury in this pathology. Furthermore, we try to dissect the molecular mechanism(s) by which displays its remarkable protective action dexrazoxane (ICRF-187), the only approved cardioprotective agent in clinical practice. Traditionally, dexrazoxane has been thought to act through its metal-chelating hydrolysis product ADR-925, which could displace iron bound to anthracycline or chelate free or loosely bound iron, thus preventing site-specific iron-based oxygen radical damage. However, recent data of both our group as well as others provided strong experimental evidence that the dexrazoxane cardioprotective effects are rather related to inhibition and/or depletion of topoisomerase IIβ enzyme.
we study the molecular mechanisms of anthracycline cardiotoxicity and particularly aim at advancement of understanding of the roles of redox-active iron, topoisomerase II and mitochodrial injury in this pathology. Furthermore, we try to dissect the molecular mechanism(s) by which displays its remarkable protective action dexrazoxane (ICRF-187), the only approved cardioprotective agent in clinical practice. Traditionally, dexrazoxane has been thought to act through its metal-chelating hydrolysis product ADR-925, which could displace iron bound to anthracycline or chelate free or loosely bound iron, thus preventing site-specific iron-based oxygen radical damage. However, recent data of both our group as well as others provided strong experimental evidence that the dexrazoxane cardioprotective effects are rather related to inhibition and/or depletion of topoisomerase IIβ enzyme.
We are also engaged in several collaborative projects aimed at search for alternative cardioprotective drug candidates with potential to differentially modulate anthracycline toxicity towards cardiac and cancer cells.

Apart from the anthracyclines, we also study cardiotoxic properties of other anticancer drugs, such as the tyrosine kinase inhibitor sunitinib or proteazome inhibitor bortezomib – both alone as well as in combinations with the anthracyclines.
<<< back on top
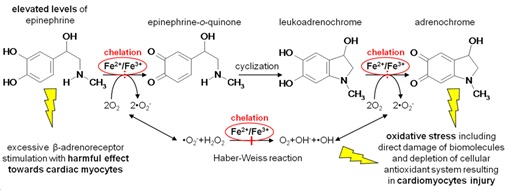
Reactive oxygen species (ROS)-related damage has been implicated in various drug toxicities, ischemia/reperfusion tissue injury, inflammation, as well as in a variety of other conditions and diseases. Myocardial tissue which is predominantly composed of terminally differentiated cardiomyocytes has very limited (if any) regenerative capacity. Hence, it is particularly vulnerable to various types of damage and cardiomyocyte injury mostly results in an irreversible impairment of the myocardial structure and function. Despite 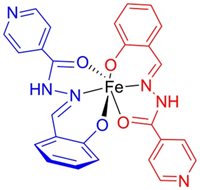 the well-known role of free radicals, conventional treatment of cardiovascular disease lacks the use of antioxidants due to negative study outcomes. Unfortunately, efforts to reduce toxicity of oxidative stress have traditionally focused primarily on the use of ROS scavengers, rather than on actual prevention of ROS production. The latter can be achieved with iron-chelating agents. Free cellular iron ions catalyze the Fenton reaction, yielding highly reactive and toxic hydroxyl radicals. Iron chelation therefore represents an attractive means of cardioprotection. Results from our laboratory as well as other groups have shown promising cardioprotective potential of salicyladehyde isonicotinoyl hydrazone (SIH) as well as some other cell membrane-permeable chelators both in vitro and in vivo. Apart from various model oxidative stress-inducing agents (such as the hydrogen peroxide or tert-butyl hydroperoxide) and anthracyclines, we have recently shown SIH as capable to provide efficient protection of cardiomyocytes against the toxicity of catecholamines –both by inhibition of deleterious catecholamine oxidation to reactive intermediates and prevention of ROS-mediated cellular injury.
the well-known role of free radicals, conventional treatment of cardiovascular disease lacks the use of antioxidants due to negative study outcomes. Unfortunately, efforts to reduce toxicity of oxidative stress have traditionally focused primarily on the use of ROS scavengers, rather than on actual prevention of ROS production. The latter can be achieved with iron-chelating agents. Free cellular iron ions catalyze the Fenton reaction, yielding highly reactive and toxic hydroxyl radicals. Iron chelation therefore represents an attractive means of cardioprotection. Results from our laboratory as well as other groups have shown promising cardioprotective potential of salicyladehyde isonicotinoyl hydrazone (SIH) as well as some other cell membrane-permeable chelators both in vitro and in vivo. Apart from various model oxidative stress-inducing agents (such as the hydrogen peroxide or tert-butyl hydroperoxide) and anthracyclines, we have recently shown SIH as capable to provide efficient protection of cardiomyocytes against the toxicity of catecholamines –both by inhibition of deleterious catecholamine oxidation to reactive intermediates and prevention of ROS-mediated cellular injury.
The major limitation of SIH has been its labile hydrazone bond that makes it prone to rapid plasma hydrolysis. Hence, in order to improve the hydrazone bond stability and cytoprotective properties, we develop and study novel ketone SIH analogs with various electron donors or acceptors in the phenyl ring:
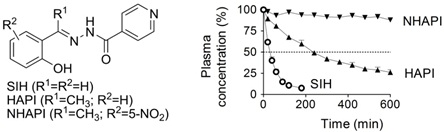
Furthermore, the use of iron chelating agents in states without systemic iron overload is known to possess the risk of toxicity due to the iron depletion. We therefore recently started to study novel "masked" iron pro-chelators that have little or no affinity for iron until the mask is selectively removed by the action of reactive oxygen species.
<<< back on top
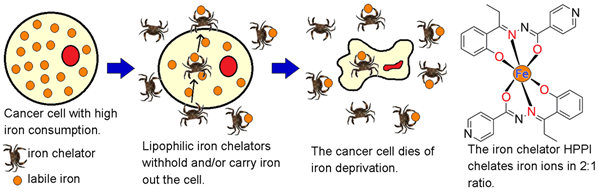
Highly proliferating cancer cells are characterized by markedly increased requirement for iron. Iron withdrawal has been in previous studies shown as an effective means of selective inhibition of tumor growth both in vitro and in vivo. We therefore participate in projects aimed at synthesis and initial preclinical assessments of novel antiproliferative aroylhydrazone and thiosemicarbazone iron chelators. Attention is here paid at selective targeting of malignant cancer cells with as little as possible interference with normal cells, including the cardiomyocytes. Apart from the effort to develop new potential anticancer drug candidates, our experiments should contribute to the understanding of molecular mechanisms by which iron depletion induces the selective anticancer effect, including the roles of cell cycle dysruption, mitochondrial injury, apoptosis or formation of free oxygen radicals. Furthermore, as the anticancer chemotherapy is often based on the combining two or more chemotherapeutic agents, we assess combinations of the newly developed agents with established drugs in search for potential synergy by disrupting different stages of the cancer cell cycle.